Research projects
Computational analysis of the relationship between histone modifications and gene expression
Rosa Karlić, Maja Kuzman, Kristian Vlahoviček
This project studies histone modifications, or modifications of proteins which participate in packing of DNA and are closely linked to gene regulation. There is evidence that histone modifications are in fact a mean for coding for certain processes downstream of genes. Quantative models for predictions of gene expression levels through histone modification have shown thus far that gene expression and histone modification have a high correlation and that a relatively small number of histone modifications are necessary to predict gene expression levels, as well as that their relationship is general and valid in many cell types.
A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nature Genetics, 2017.
Analysis of somatic microsatellite indels identifies driver events in human tumors. Nature biotechnology, 2017.
Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature, 2015.
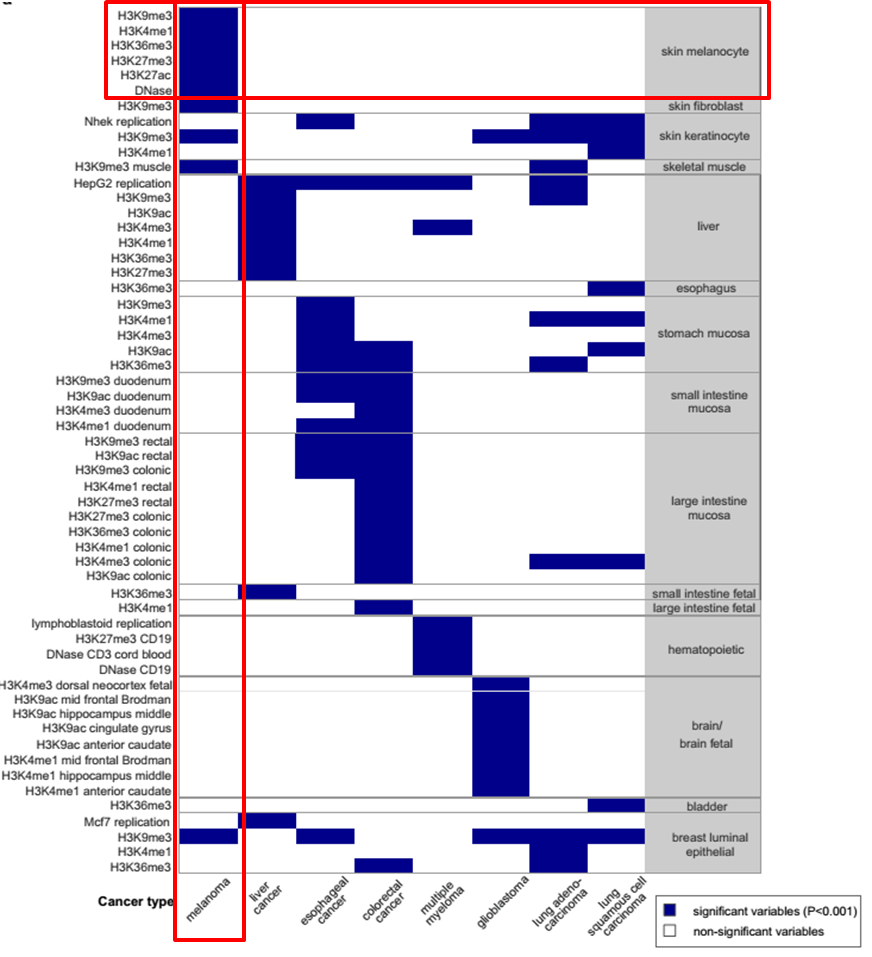
Integrative Analysis of 111 Reference Human Epigenomes. Nature, 2015.
Genetic variation in human DNA replication timing. Cell, 2014.
Histone modification levels are predictive for gene expression. PNAS, 2010.
Genomic complexity of the simplest Metazoa, the sponge
Dunja Glavaš, Juan Antonio Ruiz Santiesteban, Maja Kuzman, Kristian Vlahoviček
Sponges are living fossils, inhabiting the planet for 580 million years, and the closest living species to the first multicellular animal. Therefore, by researching sponges we are also investigating our own evolutionary past. Our research of sponge genomes has shown that, despite the morphological simplicity of sponges, their genetic repertoire is surprisingly complex. This shows that even ancient ancestors of animals already had the potential for more complex life functions. The focus in evolutionary studies is shifting from the increase of genome complexity to adaptations which enable exploitation of the existing complexity.
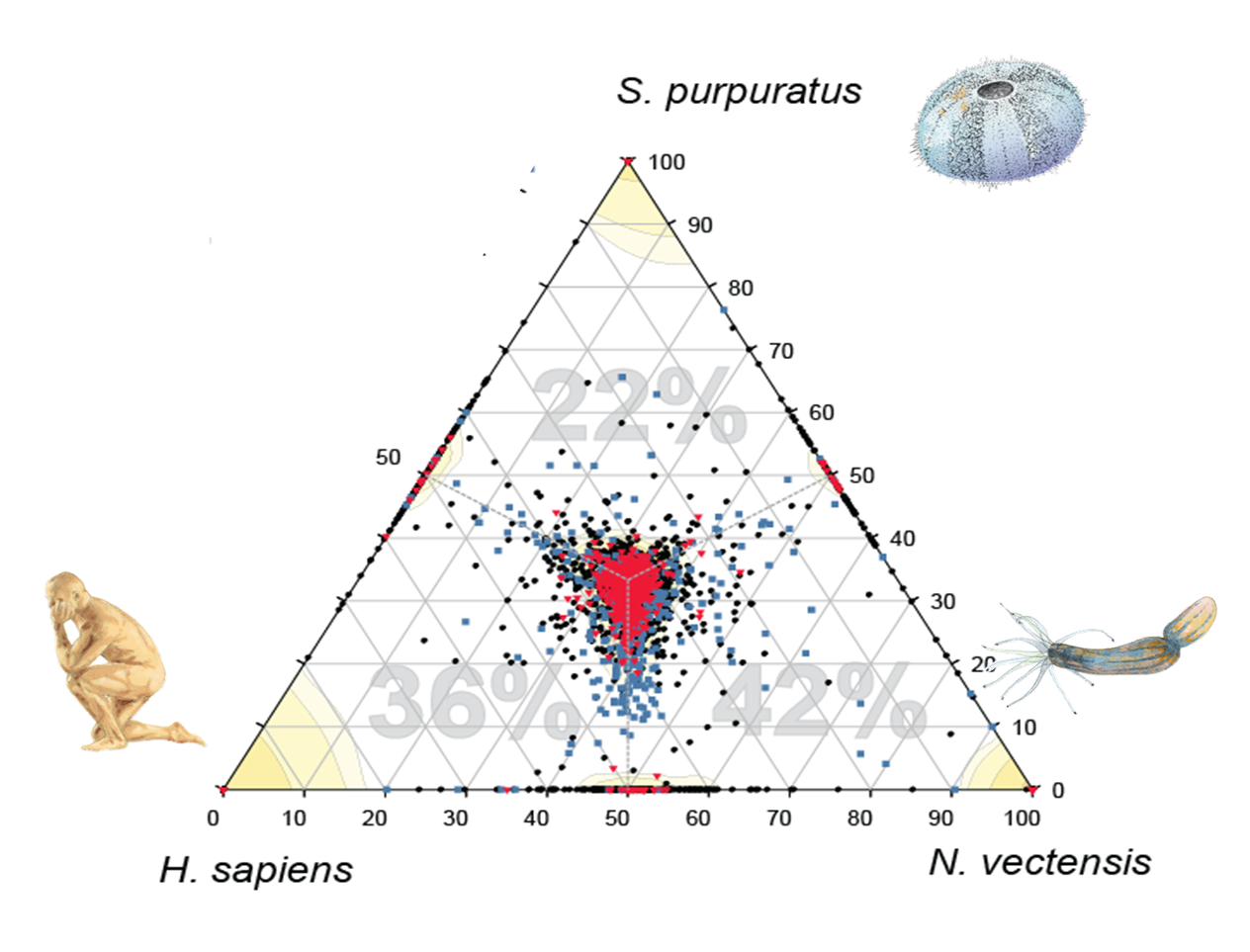
Demosponge EST sequencing reveals a complex genetic toolkit of the simplest metazoans. Molecular Biology and Evolution, 2010.
Over-represented localized sequence motifs in ribosomal protein gene promoters of basal metazoans. Genomics, 2011.
Codon usage in metagenomes
Eva Pavlinek, Maja Kuzman, Kristian Vlahoviček
Metagenomics, the study of bacterial communities isolated straight from the environment, is a field which investigates bacterial populations in their natural environments. We identify functional adaptations of whole bacterial communities which enable them to survive in diverse environmental conditions such as animal digestive tracts, where bacteria degrade food into forms which can be harvested by the animal and where changes in bacterial composition can lead to obesity and disease. The method we are developing can not only identify the functions bacteria posses but also predict how much they are used and therefore offer a more accurate description of life in bacterial communities.
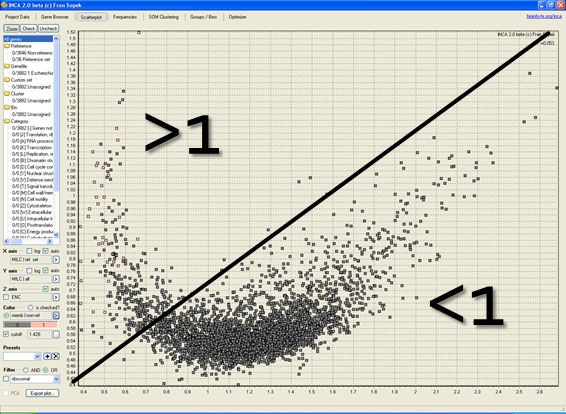
Big Data, Evolution, and Metagenomes: Predicting Disease from Gut Microbiota Codon Usage Profiles. Methods in Molecular Biology, 2016.
Environmental shaping of codon usage and functional adaptation across microbial communities. Nucleic Acids Research,2013.
Translational Selection Is Ubiquitous in Prokaryotes. PLOS Genetics, 2010.
Comparison of codon usage measures and their applicability in prediction of microbial gene expressivity. BMC Bioinformatics, 2005.
Role of long non-coding RNAs during OET
Maja Kuzman, Rosa Karlić, Kristian Vlahoviček, in collaboration with Petr Svoboda from Institute of Molecular Genetics of the ASCR
Retrotransposons are “copy-and-paste” insertional mutagens that substantially contribute to mammalian genome content. Retrotransposons often carry long terminal repeats (LTRs) for retrovirus-like reverse transcription and integration into the genome. Long non-coding RNAs (lncRNAs) are a heterogeneous group of genome-encoded RNAs, many of which have been shown to have important biological functions. With our collaborators from the Laboratory of epigenetic regulations, group of Petr Svoboda we generated a catalogue of lncRNAs expressed during oocyte-to-embryo transition from microarray and next generation sequencing datasets. Remarkably, many of the identified lncRNAs are novel and have unique expression patterns.
Long non-coding RNA exchange during the oocyte-to-embryo transition in mice. DNA Research, 2017.
Long terminal repeats power evolution of genes and gene expression programs in mammalian oocytes and zygotes. Genome Research, 2017.
The first murine zygotic transcription is promiscuous and uncoupled from splicing and 3’ processing. The EMBO Journal, 2015.
A retrotransposon-driven Dicer isoform directs endogenous siRNA production in mouse oocytes. Cell, 2013.
Contributions of nuclear topology and chromatin factors to HIV integration site selection
Maja Kuzman, Kristian Vlahoviček, in collaboration with Marina Lusic from Universitätsklinikum Heidelberg
Human immunodeficiency virus (HIV-1) has the capacity to establish latent infections, which is a major barrier to the development of a cure for AIDS. To ensure productive infection, HIV-1 has to integrate into cellular DNA. The integration is not random, HIV-1 targets preferentially the outer shell of the CD4+ T cell nucleus and it has been shown that nuclear 3D structure plays a role in dictating HIV-1 integration patterns. In collaboration with group of Marina Lusic, we are now focused to better understand what defines genomic regions that are recurrently targeted by HIV-1 and how cromatin features and their 3D nuclear organisation influences HIV-1 integration pattern selection.
Spatially clustered loci with multiple enhancers are frequent targets of HIV-1. bioRxiv, 2018.